Pitiriasis rubra pilaris
| ||||
Definición
Es una enfermedad crónica, caracterizada por placas rojoanaranjadas, escamosas y pápulas queratósicas foliculares. Se presenta de 2 formas: familiar y adquirida.
Aspecto histórico
El primer caso fue descrito por Alandius Tarral, en 1928; el nombre pitiriaris rubra pilaris fue dado a esta enfermedad por Besnier, en 1889.
Incidencia
La enfermedad no es rara, pero su incidencia no ha sido reportada con exactitud. Ambos sexos se afectan por igual. El tipo familiar ocurre tempranamente en la infancia.
Genética
El tipo familiar tiene una herencia autosómica dominante. Los pacientes que desarrollan tardíamente esta enfermedad no tienen historia familiar de afectados.
Etiología y patogenia
No están bien definidas, se ha descrito que el déficit de vitamina A o su anormal metabolismo desempeña un papel en su causa. Esto se basa en estudios realizados por Frazier y Hu, al detectar pacientes con carencia de esta vitamina, los cuales tenían lesiones en la piel, sugerentes de pitiriasis rubra.
Histopatología
La hiperqueratosis es la alteración más común. Se presenta paraqueratosis alrededor de los orificios foliculares. Hay acantosis y degeneración irregular de la capa basal; puede acompañarse de un ligero infiltrado crónico de la dermis superior.
Cuadro clínico
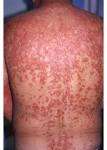
Al comienzo de la enfermedad se presenta enrojecimiento y descamación en la cara y el cuero cabelludo. Posteriormente hay una aparición de pápulas foliculares que son características de la enfermedad, y que tienen apariencia de un tapón hiperqueratósico rodeado por eritema. Con estas lesiones surge una erupción diseminada o generalizada en forma de placas de color rojonaranja, con escamas que varían desde muy finas hasta gruesas; las escamas más gruesas generalmente se observan en palmas y plantas, y ocasionan fisuras. Las uñas cambian de color, se tornan amarillo carmelitosas con hiperqueratosis subungueal; la lámina se debilita y se presentan puntos hemorrágicos. En las membranas mucosas pueden aparecer placas blancolechosas y erosiones. El pelo y los dientes son normales. El prurito es poco frecuente. Las manifestaciones sistémicas no están presentes, excepto en los casos generalizados que pueden evolucionar a una eritrodermia.
Diagnóstico
Una historia familiar de la enfermedad, acompañada de la presencia de hiperqueratosis folicular, con dermatitis escamosa y la presencia de islotes de piel normal, hacen el diagnóstico, el cual se corrobora con la biopsia.
Diagnóstico diferencial
Se puede confundir con psoriasis en las fases iniciales de la enfermedad.
Tratamiento
En el tratamiento se deben seguir las recomendaciones siguientes:
Recomendaciones generales
Se debe realizar consejo genético.
Tratamiento sistémico
La terapia con retinoides resulta muy efectiva en esta entidad; se utiliza el isotretinoin en dosis de 0,5-1 g/kg/día; vitamina A, en dosis de 1x10E6 UI / día, durante 14 días; el methotrexate puede ser utilizado, con beneficios en algunos pacientes.
Tratamiento tópico
Uso de soluciones queratolíticas que contienen propilenglicol y ácido láctico en apósitos plásticos oclusivos de 2 a 4 horas, seguido de un aceite esteroideo de 4 a 8 horas, también de forma oclusiva.
Curso y pronóstico
Esta enfermedad es crónica y persistente. El paciente se afecta grandemente desde el punto de vista psicológico; en ocasiones se ha reportado suicidios.
Fuentes
- Colectivo de autores. Dermatología. Editorial Ciencias Médicas, 2002. pág 53.